Taxonomic Classification - Centrifuge
Centrifuge
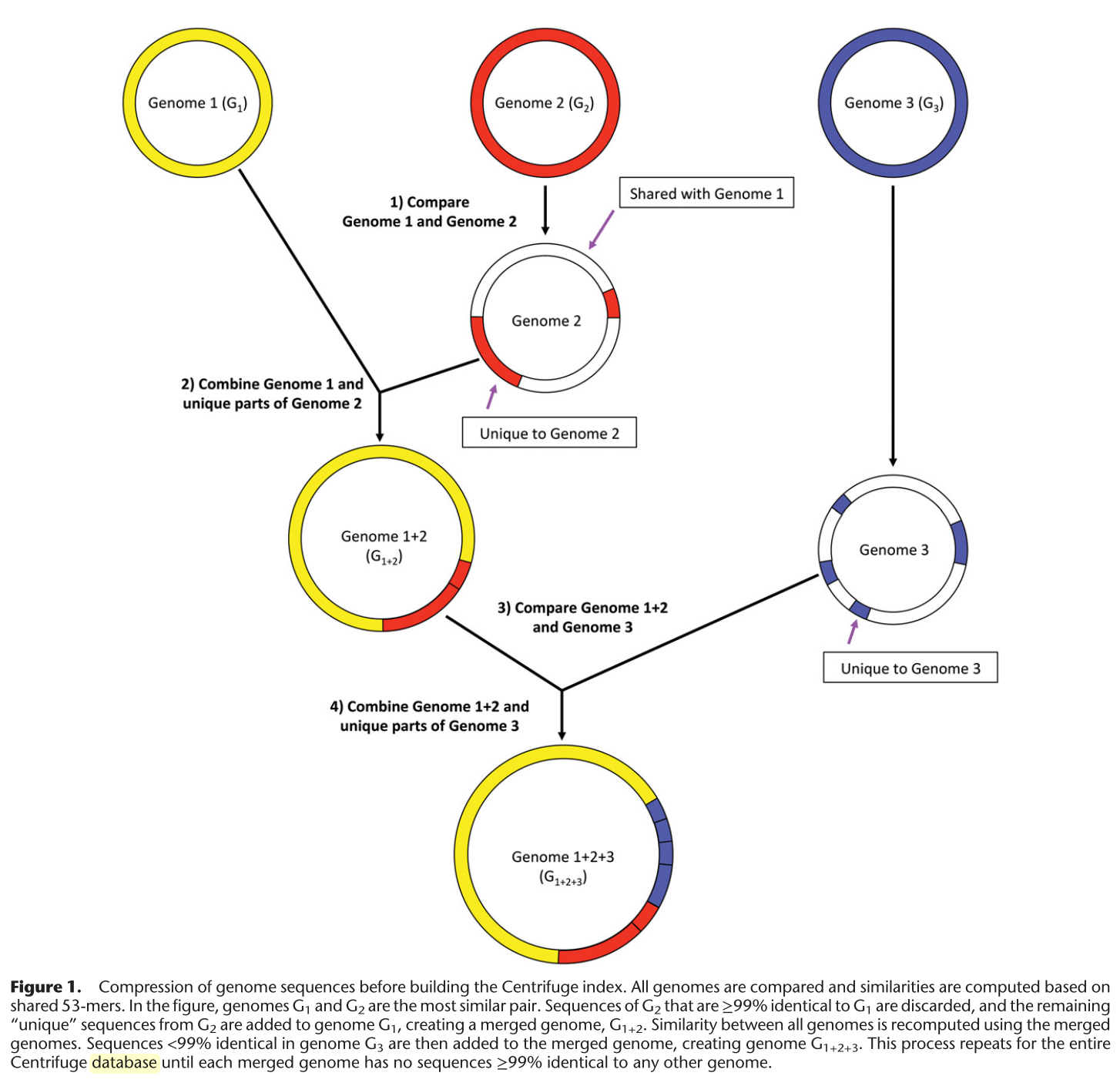
- Create an FM-Index of the reference sequence.
-
Uses a BWA to align reads to an FM-index of reference genomes. Outputs the matches. If the number of matches is large, the algorithm will climb the phylogenetic tree to assign a higher phylo instead.

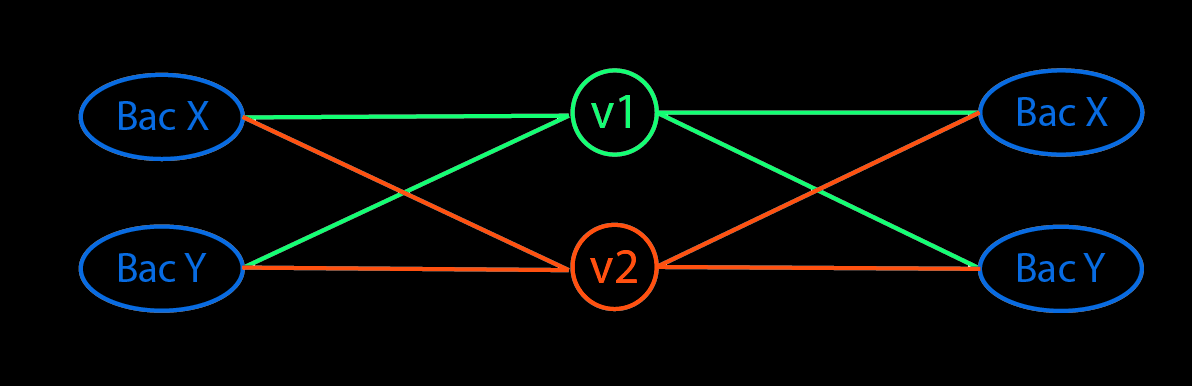
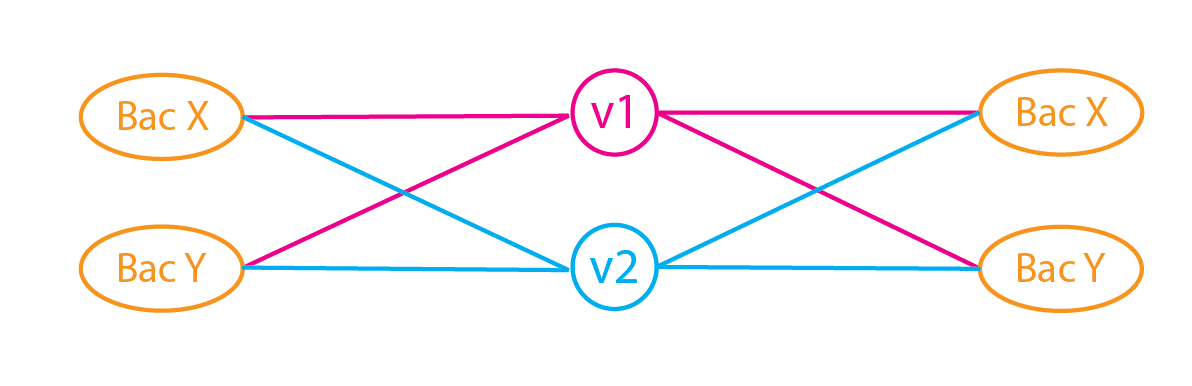
- Represent a match between read $i$ and reference genome $j$ with an indicator $C_{ij}$. $C$ is an indicator matrix.
- To find the relative abundance of species, use an EM algorithm. The algorithm is roughly as follow:
- Since each read (likely) corresponds to multiple reference genomes, we can assign a probability that the read came from a genome. Set the prob to be proportional to the relative abundance (currently unknown) of the corresponding species.
- Guess the initial abundances $\alpha_j$.
-
Create a probability matrix $P$ by multiplying $C$ with $\alpha$ and normalizing the values so that each row sum to 1 i.e. the total probability that a read come from the set of genome is 1.
Reads\Genome G1 G2 G3 G4 R1 $P_{11}=\overline{C_{11}*\alpha_1}$ $\overline{C_{12}*\alpha_2}$ $\overline{C_{13}*\alpha_3}$ $\overline{C_{14}*\alpha_4}$ R2 $\overline{C_{21}*\alpha_1}$ $\overline{C_{22}*\alpha_2}$ $\overline{C_{31}*\alpha_3}$ $\overline{C_{41}*\alpha_4}$ Sum $n_1$ $n_2$ $n_3$ $n_4$ - Sum each column. Each sum corresponds to the total (probabilistic) reads assigned to each sequence ($n_j$).
- To calculate a new estimate for the relative abundance of species $j$, normalize $n_j$ by the total read count. \(\alpha_j=\frac{n_j}{\sum_jn_j}\) This estimate works assuming that the genome lengths are equal across all species. To account for genomes of different length, divide $n_j$ by genome length $l_j$ and normalize that instead. \(\alpha_j=\frac{n_j/l_j}{\sum_jn_j/l_j}\)
A note on genome length. Centrifuge calculates the mean of all sequences assigned to a particular taxID based on NCBI’s taxonomic database. In other words, if there are short partial sequences in the database, Centrifuge assumes they are full genomes and uses them in the averaging as is. For example, the genome size of taxID 590 (Salmonella genus) is 202 because the only sequence in the ncbi nt 2008 database assigned to 590 is a Salmonella repetitive sequence of 202 bp. A carefully curated database is required for Centrifuge abundance estimation step to be effective.
You can use centrigue-inspect to extract the genome sizes (via size table), seqID to taxID (via conversion table), raw sequences. Use seqtk to extract sequences based on seqID, and count the basepair of sequences assigned to a particular taxID. It should match the number in the size table.